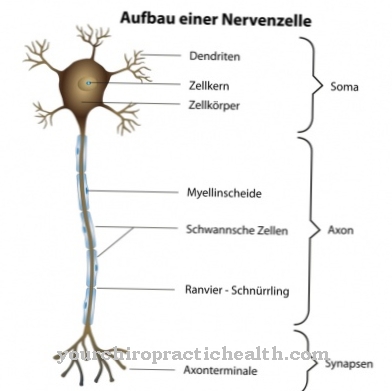
Itu Sindrom Bardet-Biedl, juga Sindrom Laurence-Moon-Biedl-Bardet (LMBBS), adalah penyakit dari bidang ciliopathies yang terjadi secara eksklusif karena faktor keturunan. Sindroma tersebut memanifestasikan dirinya dalam bentuk beberapa malformasi yang dipicu oleh perubahan (mutasi) pada lokasi gen atau kromosom yang berbeda.
Apa itu Sindrom Bardet-Biedl?

© Gambar Creativa - stock.adobe.com
Gambaran klinis yang ditentukan oleh dokter Moon dan Laurence dan kemudian oleh Bardet dan Biedl adalah penyakit di mana distrofi retina muncul sebagai gambaran medis yang signifikan dalam kombinasi dengan gejala lain. Karena situasi awal medis yang rumit ini, penentuan akhir penyakit BBS menjadi sulit. Gambaran klinis ini dicatat secara medis untuk pertama kalinya pada tahun 1866.
Empat orang yang diperiksa menderita retinitis pigmentosa (distrofi retinal, RP) sehubungan dengan paraplegia (kelumpuhan spastik) serta hipogenitalisme (organ genital yang kurang berkembang) dan cacat mental. Pada tahun 1920, dokter Prancis Bardet menjelaskan penyakit yang terdiri dari RP (distrofi retinal), hipogenitalisme, polidaktili, dan obesitas.
Ahli patologi Praha Biedl juga menemukan kelemahan (kebingungan mental). Pada tahun 1925, peneliti Weiss dan Solis-Cohen meringkas kasus yang diketahui dan menggambarkan gambaran klinis sebagai Sindrom Laurence-Moon-Biedl-Bardet.
penyebab
Di tahun-tahun berikutnya, literatur medis semakin menunjukkan bahwa kasus yang dicatat oleh Laurence dan Moon adalah bentuk khusus langka yang hanya terjadi pada kasus individu bersama dengan BBS. Hasil penelitian medis yang lebih baru menetapkan sindrom Bardet-Biedl ke area ciliopathies (penyakit ciliary).
Penyakit ini menunjukkan kerusakan umum dari apa yang disebut silia (proses kecil, antena), yang terjadi pada sebagian besar sel organisme manusia. Ciliopathies ditandai dengan transisi yang mengalir dan tumpang tindih antara penyakit silia yang berbeda.
Gejala, penyakit & tanda
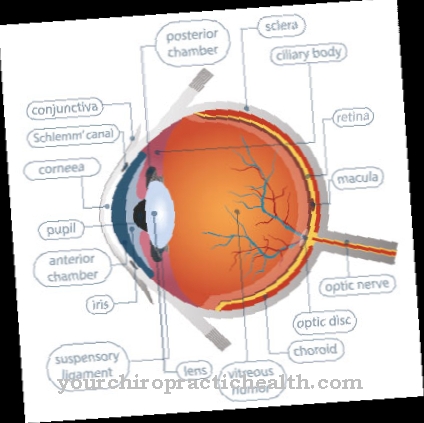
Ciri utama dari distrofi retinal herediter adalah istilah umum yang menggambarkan onset hilangnya fungsi dan degenerasi (penghancuran) fotoreseptor. Mereka menyebabkan hilangnya fungsi visual secara progresif (progresif). Gangguan penglihatan yang berkembang pesat biasanya muncul sangat dini pada anak-anak ketika mereka berusia antara empat dan sepuluh tahun. Mereka membuat diri mereka terasa dengan cara yang berbeda, tergantung pada fotoreseptor yang terpengaruh.
Sebagai "bentuk kerucut batang" dengan perjalanan karakteristik retinitis pigmentosa (RP), penyakit ini berasal dari perifer retina (retina luar) dan berkembang menjadi degenerasi makula (kerusakan penglihatan yang tajam) melalui hilangnya bidang pandang secara progresif.
Dengan obesitas (obesitas), tubuh menunjukkan akumulasi patologis jaringan lemak. Dalam kasus BBS, penumpukan lemak yang meningkat secara tidak normal di kaki, perut, bokong, lengan, dada dan pinggul terutama terjadi sebagai obesitas batang, dengan batang tubuh, tungkai dan paha menjadi sangat terpengaruh. Polydactyly adalah gejala yang terlihat dan ciri signifikan dari sindrom Bardet-Biedl. Penemuan ini tidak mudah karena polidaktili rudimenter dikoreksi secara pembedahan setelah lahir.
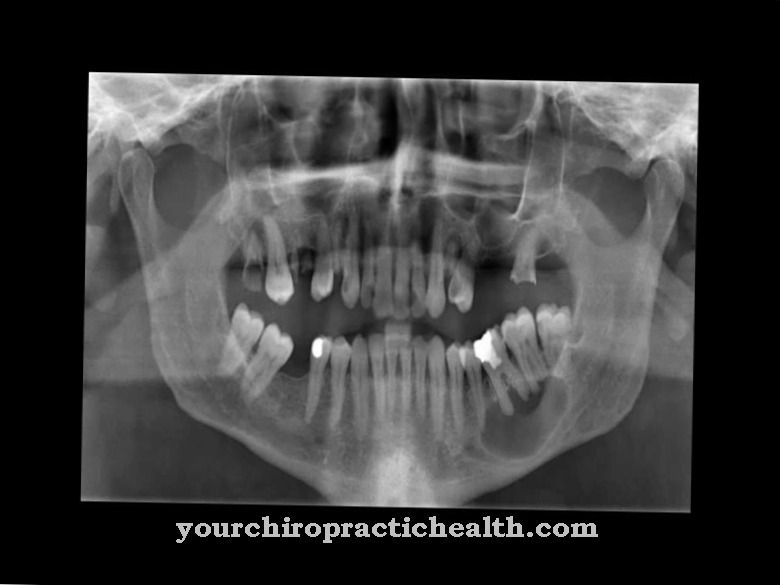
Sinar-X dapat memberikan informasi lebih lanjut. Polydactyly dapat muncul dengan tanda yang berbeda, misalnya sebagai pelengkap jari kaki atau jari yang belum sempurna. Jari kaki atau jari dapat dibentuk sebagai tambahan atau hanya sebagian. Hexadactyly unilateral pada kaki dan / atau tangan memiliki link tambahan, hexadactyly bilateral terjadi pada kedua kaki dan / atau tangan.
Jari kaki atau jari yang tumbuh bersama (sindaktili) dan pemendekan satu atau lebih jari kaki atau jari (brachydactyly) juga merupakan tanda BBS. Hanya sedikit pasien yang memiliki keempat ekstremitas yang terkena. Penundaan perkembangan mental berbeda. Hanya sejumlah kecil dari mereka yang terkena menunjukkan keterbelakangan mental yang parah. Kecerdasan yang biasanya terlatih adalah mungkin.
Anak-anak belajar berbicara dan berjalan terlambat, dan terkadang mereka menunjukkan masalah perilaku seperti gangguan kecemasan. Perilaku kompulsif atau autis, ambang batas rendah untuk frustrasi, dan emosi yang tidak stabil adalah kemungkinan efek samping lain. Familiar lebih disukai, tetapi perubahan ditolak. Kelainan pada organ genital internal dan eksternal sering terjadi.
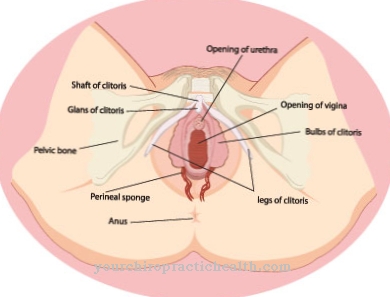
Perubahan lainnya adalah hipospadia (pembukaan uretra di atas atau di bawah, bukan di depan penis), testis perut atau inguinal, konstriksi uretra, penyempitan kulup dan katup uretra posterior. Pada pasien wanita, atresia vagina (vagina tidak terbuka), lubang uretra hilang dan labia bagian dalam berkurang diketahui.
Tidak jarang wanita yang terkena mengalami siklus menstruasi yang tidak teratur. Perubahan ginjal adalah efek samping yang umum. Temuannya tergantung dari pemeriksaan saluran kemih bagian bawah dan ginjal menggunakan USG (sonografi).
Diagnosis & perjalanan penyakit
Sindrom Bardet-Biedl (BBS) memiliki enam gejala utama, tetapi tidak muncul bersamaan dalam setiap kasus. Dokter mengasumsikan temuan yang sesuai jika setidaknya ada empat gejala utama yang muncul. Selain itu, kemungkinan besar penyakit muncul jika pasien memiliki tiga gejala utama dan dua gejala sekunder.
Enam gejala utama adalah distrofi retina, obesitas (akumulasi jaringan adiposa yang tidak normal, kelebihan berat badan), polidaktili (jari kaki dan / atau jari berlebih), keterbelakangan mental (keterlambatan perkembangan mental), hipogenitalisme (organ genital yang kurang berkembang) dan penyakit ginjal. Gejala sekunder frekuensi rendah termasuk keterlambatan bicara, defisit bicara, malformasi jantung, ataksia (gangguan koordinasi gerakan), asma, diabetes mellitus (diabetes), penyakit Crohn (radang usus besar dan / atau kecil), displasia tulang rusuk dan tulang belakang, dan kyphoscoliosis (anemia vertebral) di.
Komplikasi
Dengan sindrom Laurence-Moon-Biedl-Bardet, mereka yang terkena biasanya menderita kehilangan fungsi visual. Kerugian tidak terjadi secara tiba-tiba, melainkan bertahap. Dalam kasus terburuk, mereka yang terkena akan menjadi buta total, yang biasanya tidak dapat diobati lagi.
Khususnya pada remaja dan anak-anak, kebutaan bisa menimbulkan keluhan psikologis yang parah atau bahkan depresi. Para pasien jelas dibatasi dalam kehidupan sehari-hari mereka dan menderita bidang penglihatan yang sangat berkurang. Dalam banyak kasus, sindrom Laurence-Moon-Biedl-Bardet juga menyebabkan masalah perilaku, sehingga anak-anak khususnya dapat mengalami perundungan atau pelecehan.
Perkembangan anak-anak juga sangat tertunda dan dibatasi oleh sindrom tersebut. Gangguan kecemasan juga bisa terjadi. Tidak jarang sindrom Laurence-Moon-Biedl-Bardet menyebabkan keluhan psikologis dan depresi pada kerabat atau orang tua. Sayangnya, pengobatan kausal untuk sindrom Laurence-Moon-Biedl-Bardet tidak memungkinkan.
Beberapa keluhan bisa dibatasi. Namun, perjalanan penyakit yang sepenuhnya positif tidak terjadi. Sindroma tersebut tidak mengurangi harapan hidup pasien. Dalam beberapa kasus, mereka yang terkena dampak terkadang membutuhkan bantuan dari orang lain dalam kehidupan sehari-hari mereka.
Kapan sebaiknya Anda pergi ke dokter?
Karena sindrom Laurence-Moon-Biedl-Bardet adalah penyakit keturunan, diagnosis dapat dibuat di dalam rahim. Paling lambat setelah melahirkan, dokter harus dikonsultasikan jika ada gejala khas seperti gangguan penglihatan atau obesitas. Malformasi pada jari kaki dan jari juga merupakan indikator yang jelas dari suatu penyakit.Orang tua yang memperhatikan gejala pada anak mereka harus segera memberi tahu dokter anak.
Pemeriksaan komprehensif memberikan informasi tentang penyakit tersebut. Setelah itu, terapi biasanya dimulai secara langsung, yang terdiri dari berbagai perawatan oleh ahli ortopedi, ahli saraf, dokter mata, ahli penyakit dalam dan terapis serta fisioterapis. Kunjungan lebih lanjut ke dokter diperlukan jika pengobatan tidak memberikan efek yang diinginkan. Nasihat medis juga diperlukan dalam situasi darurat, misalnya jika anak jatuh akibat kelainan bentuk atau tiba-tiba mengalami kejang. Jika orang yang sakit menunjukkan tanda-tanda ketidaknyamanan emosional, orang tua harus berkonsultasi dengan terapis yang sesuai. Anak-anak yang lebih besar dapat menghubungi psikolog sekolah bersama dengan orang tua mereka dan mendiskusikan tindakan yang sesuai.
Terapi & Pengobatan
Penyakit ini terjadi atas dasar pewarisan resesif autosom, yang berarti bahwa kedua salinan (alel) gen BBS menunjukkan perubahan (mutasi). Orang tua pasien adalah "berdarah campuran" dan masing-masing membawa alel yang dimodifikasi dan tidak berubah dari gen yang sesuai. Mereka tidak mengidap penyakit itu. Anak-anak hanya akan sakit jika ayah dan ibunya mewariskan alel yang bermutasi. Dengan tambahan anak, kemungkinan pengulangan adalah 25 persen.
Pilihan terapi kausal belum diketahui, karena gejala penyakit tertentu belum dapat secara pasti dikaitkan dengan berbagai perubahan genetik. Gejala dan manifestasinya tampak berbeda bahkan pada saudara kandung yang sakit. Karena gambaran lengkap karakteristik BBS hanya hadir dalam kasus yang jarang terjadi, terutama pada anak kecil, diagnosis yang sesuai sulit dilakukan.
Karena gejala oligosimtomatik yang sering muncul, dengan gejala atipikal yang sangat sedikit dan hanya sedikit timbul, gambaran klinis lain yang mungkin harus diperhitungkan dalam diagnosis banding. Perubahan gen yang sama dapat menyebabkan gambaran klinis yang berbeda, misalnya sindrom Joubert, Bardet-Biedl atau Meckel-Gruber.
Outlook & ramalan
Prognosis adanya sindrom Laurence-Moon-Biedl-Bardet umumnya buruk karena malformasi multipel bersifat bawaan dan tidak dapat disembuhkan. Jika empat dari enam gejala utama terjadi, diagnosis sindrom Laurence-Moon-Biedl-Bardet dipastikan. Berbagai gejala sekunder menambah gejala utama. Ini termasuk kebutaan yang merayap.
Karena kompleksitas gejala tidak ada kemungkinan untuk sembuh. Hanya ada kemungkinan biasa-biasa saja untuk meredakan gejala yang terlihat. Jumlah kemungkinan malformasi dan kelainan pada sindrom Laurence-Moon-Biedl-Bardet sangat besar sehingga penyakit keturunan sulit untuk diobati. Bagaimanapun, perjalanan penyakit genetik ini tidak dapat dipengaruhi. Namun, gejala saat ini dapat dikurangi sebagian.
Namun, prognosis keseluruhan yang buruk tidak mengurangi harapan hidup orang yang terkena. Pada usia lanjut dan setelah menjadi buta, mereka yang terkena dampak dapat secara permanen bergantung pada bantuan atau perawatan. Melalui upaya medis interdisipliner, banyak penderita sindrom Laurence-Moon-Biedl-Bardet dapat mengalami perjalanan penyakit yang lebih ringan.
Masalah penglihatan yang meningkat merupakan bagian dari penyakit yang sulit diobati dan bermasalah.Peningkatan gangguan penglihatan sudah terjadi pada anak kecil yang terkena. Mereka memburuk seiring waktu. Masalah penglihatan tidak harus menyebabkan kebutaan pada semua yang terpengaruh. Gejala sisa psikologis dari sindrom Laurence-Moon-Biedl-Bardet biasanya dapat diobati dengan baik.
pencegahan
Pencegahan dalam arti mencegah penyakit ini tidak mungkin dilakukan. Pemantauan gejala secara teratur dan gejala yang menyertainya adalah penting. Pemeriksaan tekanan darah dan fungsi ginjal berulang kali, nasihat nutrisi, fisioterapi dan terapi okupasi serta terapi wicara adalah pendekatan terapeutik yang memungkinkan.
Rehabilitasi
Dalam kebanyakan kasus, mereka yang terkena sindrom Laurence-Moon-Biedl-Bardet tidak memiliki pilihan tindak lanjut khusus yang tersedia, sehingga dokter harus dihubungi dan dikonsultasikan sejak dini pada penyakit ini. Biasanya, penyembuhan diri tidak dapat terjadi, jadi perawatan oleh dokter selalu diperlukan.
Karena sindrom Laurence-Moon-Biedl-Bardet merupakan penyakit keturunan, maka orang yang bersangkutan harus menjalani pemeriksaan genetik dan dilakukan nasehat jika ingin memiliki anak agar sindrom Laurence-Moon-Biedl-Bardet tidak diturunkan kepada keturunannya. diteruskan. Dalam banyak kasus, mereka yang terkena bergantung pada intervensi bedah untuk mengurangi malformasi dan deformitas.
Di sini, orang yang terkena dampak harus beristirahat setelah prosedur dan merawat tubuhnya. Pengerahan tenaga atau aktivitas fisik dan stres lainnya harus dihindari dalam hal apa pun agar tidak membebani tubuh secara tidak perlu. Karena sindrom Laurence-Moon-Biedl-Bardet juga dapat menyebabkan perilaku abnormal, orang tua harus mendukung dan mendorong anak dalam perkembangannya. Diskusi yang penuh kasih dan intensif dengan anak juga diperlukan untuk mencegah gangguan psikologis atau depresi.
Anda bisa melakukannya sendiri
Sindrom Laurence-Moon-Biedl-Bardet memiliki berbagai gejala, dengan pasien yang paling sering menderita gangguan fungsi penglihatan. Bahkan pada anak-anak, kemampuan melihat yang biasa mulai menurun, sehingga orang tua yang menyerahkan anak tersebut ke dokter dan dengan demikian mempercepat diagnosis. Dengan cara ini, penyakit dapat diobati dengan cepat, meskipun pilihan pengobatan sejauh ini hanya bersifat simptomatis.
Gangguan penglihatan semakin meningkat pada anak yang sakit sehingga sangat mengganggu kehidupan sehari-hari, sehingga kualitas hidup mereka yang terkena semakin menurun. Karena masalah penglihatan menimbulkan banyak kesulitan bagi pasien ketika bersekolah, di waktu senggang dan berkaitan dengan integritas fisik mereka. Risiko kecelakaan juga meningkat secara signifikan, misalnya pada lalu lintas jalan raya. Itulah sebabnya orang tua sedapat mungkin mendampingi anak mereka yang sakit atau mempekerjakan staf perawat agar pasien tidak dibiarkan mengurus dirinya sendiri.
Dalam beberapa kasus, penyakit ini menyebar hingga mencapai kebutaan. Karena perkembangan seperti itu sudah terbukti sebelumnya, pasien mempersiapkannya. Orang tua mendesain ulang ruang hidup agar tidak mengandung sumber bahaya bagi penyandang tunanetra. Selain itu, para korban tunanetra belajar cara menggunakan tongkat panjang agar dapat bergerak sendiri di luar rumah.
.jpg)








.jpg)

.jpg)