Sindrom Smith-Magenis adalah sebutan untuk penyakit keturunan yang jarang terjadi. Ini disebabkan oleh hilangnya kromosom 17.
Apa itu Sindrom Smith-Magenis?

© vchalup– stock.adobe.com
Pada Sindrom Smith-Magenis (SMS) adalah penyakit genetik yang langka. Orang yang terkena tidak memiliki bagian kecil dari kromosom 17. Namun, bagian kecil ini penting karena berisi informasi yang penting untuk pematangan sehat anak di dalam kandungan ibu.
Dua spesialis genetika Ann Smith dan Ellen Magenis adalah nama yang sama dari Sindrom Smith-Magenis. Para dokter pertama kali mendeskripsikan penyakit ini pada awal 1980-an. Menurut penelitian di Amerika, prevalensi sindrom Smith-Magenis adalah 1 dari 25.000 kelahiran. Sejauh ini, penyakit tersebut hanya muncul secara sporadis di negara-negara berbahasa Jerman. Tidak ada perbedaan antara jenis kelamin saat terjadinya sindrom ini.
penyebab
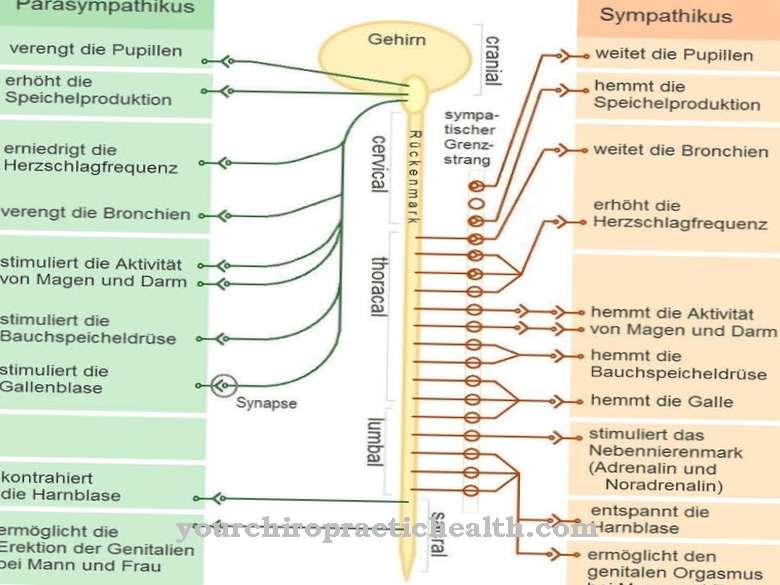
Sindrom Smith-Magenis disebabkan oleh kurangnya sebagian kecil kromosom 17, yang berarti informasi di sana juga hilang. Dalam kedokteran, kurangnya data ini disebut sebagai penghapusan 17p11.2. P adalah singkatan dari "petit", yang berarti "kecil" dan menunjukkan lengan kromosom yang pendek.
Berapa banyak bagian kromosom yang hilang bervariasi dari orang ke orang. Untuk alasan ini, intensitas sindrom Smith-Magenis bervariasi. Ada beberapa gen di bagian kromosom yang hilang. Namun, gejala sindrom Smith-Magenis terutama disebabkan oleh kurangnya gen RAI1.
Beberapa ahli medis berpendapat bahwa kurangnya gen lain berkontribusi pada perbedaan karakteristik penyakit individu. Studi ilmiah masih dilakukan pada hubungan yang tepat. Menurut pengetahuan ilmiah saat ini, tidak ada faktor risiko seperti pengaruh lingkungan atau kesalahan ibu selama kehamilan.
Inilah bagaimana sindrom Smith-Magenis berkembang secara spontan. Mutasi pada kromosom ke-17 terjadi secara kebetulan dan dimulai sebelum pembuahan. Mutasi dimungkinkan baik pada ibu maupun ayah. Namun, mungkin saja anak-anak dari orang tua yang terkena tetap sehat meskipun terjadi penghapusan kromosom 17 dan tidak otomatis menjadi sakit.
Gejala, penyakit & tanda
Sindrom Smith-Magenis disertai dengan sejumlah masalah perilaku yang khas. Ini termasuk kelainan perkembangan seperti gangguan perkembangan mental sedang hingga parah. Ini sebagian besar adalah keterlambatan perkembangan bahasa dan keterbelakangan perkembangan umum.
Karena sindrom Smith-Magenis merupakan sindrom dysmorphic, maka terdapat pula kelainan dismorfologi seperti brachycephaly (kepala pendek), midface hypoplasia, tangan lebar dan pendek, mata dalam, bibir atas melengkung, sudut mulut mengarah ke bawah, telinga rendah dan lebar. Akar hidung.
Selain itu, ada masalah perilaku yang tidak selalu dapat dikenali secara memadai pada anak kecil. Mereka yang terpengaruh menderita luka sendiri dengan membenturkan kepala ke dinding, menggigit jari tangan atau tangan, dan mencabut kuku jari kaki atau kuku.
Rasa sakitnya dirasa akan berkurang. Karena ritme siang-malam juga terganggu, pasien menderita gangguan tidur yang parah. Selain itu, terdapat gangguan kesehatan seperti gangguan pendengaran sensorineural atau konduktif, perawakan pendek atau skoliosis.
Selanjutnya terdapat perubahan pada mata seperti rabun jauh, strabismus atau kornea yang terlalu kecil, kelainan jantung, epilepsi, kelainan otak seperti keluhan ventrikulomegali atau kandung kemih.
Khasnya juga suara yang dalam dan kasar, kelelahan di siang hari, kadar hormon tiroid rendah dan tingkat imunoglobulin yang menurun. Selain itu, banyak penderita yang bertingkah laku seperti orang autis, tidak mampu berbicara, mengejar seragam, kehidupan rutin sehari-hari dan takut disentuh. Tidak jarang perilaku agresif bahkan melampiaskan amarah.
Diagnosis & perjalanan penyakit
Karena gejala sindrom Smith-Magenis sangat luas, mereka tidak cukup untuk diagnosis penyakit yang tepat. Ada beberapa penyakit yang menyerupai sindrom Smith-Magenis. Ini termasuk sindrom Prader-Willi. Menghitung dan melihat kromosom juga dianggap tidak cukup untuk diagnosis yang andal.
Karena itu, dokter melakukan tes genetik. Metode ini disebut uji IKAN (hibridisasi fluoresensi in situ). Dalam prosedur ini, sebagian darah diambil dari pasien untuk memeriksa sel darah jika tidak ada komponen kromosom 17 tertentu.
Diagnosis sindrom Smith-Magenis dianggap sulit. Diasumsikan bahwa keadaan ini berkontribusi pada rendahnya jumlah kasus. Tidak jarang banyak anak yang terkena dampak menerima diagnosis yang berbeda meskipun mereka menderita SMS. Ini sebagian besar adalah ADHD (attention deficit / hyperactivity disorder) atau autisme.
Sindrom Smith-Magenis dianggap tidak dapat disembuhkan. Perjalanan penyakit tergantung pada luasnya, usia diagnosis dan tindakan terapeutik yang sesuai. Beberapa pasien dapat hidup hingga lebih dari 80 tahun.
Komplikasi
Karena sindrom Smith-Magenis, pasien menderita berbagai penyakit mental dan fisik serta keterbatasan. Dalam banyak kasus, kerabat atau orang tua juga terkena sindrom ini dan membutuhkan dukungan serta pengobatan psikologis. Mereka yang terkena dampak menderita gangguan perkembangan yang parah dan masalah perilaku.
Oleh karena itu mereka membutuhkan dukungan khusus dan biasanya juga membantu dalam kehidupan sehari-hari mereka. Desakan untuk menyakiti diri sendiri juga bisa dirasakan akibat penyakit, sehingga perawatan di klinik tertutup mungkin diperlukan. Selain itu, ada gangguan tidur atau gangguan pendengaran. Perawakannya yang pendek bisa memicu perundungan atau ejekan, terutama di kalangan anak muda.
Mereka yang terpengaruh juga menderita gangguan penglihatan dan kelainan jantung. Pada kasus yang parah, serangan epilepsi juga dapat terjadi, yang dapat menyebabkan kematian. Karena gejalanya yang parah, harapan hidup biasanya sangat terbatas. Pengobatan sindrom ini murni bergejala. Sayangnya, tidak mungkin untuk sepenuhnya membatasi semua keluhan. Sayangnya, sindrom Smith-Magenis juga tidak dapat dicegah.
Kapan sebaiknya Anda pergi ke dokter?
Perawatan oleh dokter selalu diperlukan untuk sindrom Smith-Magenis. Karena ini merupakan penyakit keturunan, maka tidak dapat disembuhkan secara tuntas, sehingga yang terkena biasanya membutuhkan terapi seumur hidup untuk meringankan gejalanya. Jika yang bersangkutan berkeinginan memiliki anak, dapat juga dilakukan penyuluhan genetik agar sindrom Smith-Magenis tidak diteruskan kepada keturunannya.
Seorang dokter harus dikonsultasikan dengan sindrom ini jika orang yang bersangkutan menderita gangguan perkembangan yang parah atau menunjukkan masalah perilaku yang parah. Mereka yang terpengaruh sangat gelisah dan tidak bisa berkonsentrasi. Dalam banyak kasus, perilaku melukai diri sendiri juga merupakan indikasi sindrom Smith-Magenis dan harus selalu dievaluasi oleh dokter. Lebih lanjut, pasien sering menunjukkan keluhan mata atau kelainan jantung. Seorang dokter juga harus berkonsultasi dengan keluhan ini.
Sindrom Smith-Magenis dapat didiagnosis oleh dokter umum. Perawatan lebih lanjut kemudian dilakukan oleh spesialis terkait dan sangat tergantung pada tingkat keparahan gejala.
Perawatan & Terapi
Karena sindrom Smith-Magenis disebabkan oleh faktor genetik, maka tidak dapat disembuhkan. Untuk alasan ini, mengobati kondisi tersebut terbatas pada gejala. Terapi wicara, terapi okupasi, dan tindakan fisioterapi dianggap membantu.
Pelatihan komunikasi yang mencakup bahasa isyarat juga dianggap bermanfaat. Terlepas dari semua tindakan terapi, kebanyakan pasien membutuhkan bantuan sepanjang hidup mereka. Obat-obatan tertentu juga dapat digunakan untuk mengobati gejala sindrom Smith-Magenis.
Ini dapat meringankan beberapa keluhan. Pemberian hormon melatonin memperbaiki gangguan tidur. Obat lain seperti Risperidal dapat digunakan untuk melawan tindakan menyakiti diri sendiri.
pencegahan
Sindrom Smith-Magenis adalah penyakit genetik. Oleh karena itu pencegahan tidak mungkin dilakukan.
Anda bisa melakukannya sendiri
Gejala penyakit keturunan muncul segera setelah lahir. Secara alami, bayi tidak dapat memulai tindakan swadaya yang memadai.Selain itu, gangguan kesehatan sangat beragam sehingga tidak mungkin bagi orang sakit untuk memperbaiki situasi secara memadai.
Oleh karena itu, kerabat wajib merancang struktur kehidupan sehari-hari dan merawat pasien dengan cara sebaik mungkin bagi pasien. Ketika tindakan merusak diri sendiri terjadi, orang yang bersangkutan harus dilindungi dari dirinya sendiri. Jika tidak, mungkin ada komplikasi serius dan kemerosotan kesehatan. Karena kerabat sering kali kewalahan dengan perawatan pasien, perawatan yang baik harus diberikan jika perlu. Pada saat yang sama, lingkungan sosial yang stabil juga penting agar pasien tidak terkena stres tambahan.
Banyak orang sakit menderita gangguan tidur dan ritme bangun. Ini merupakan tantangan tersendiri bagi setiap orang yang terlibat, perilaku abnormal juga tidak selalu mudah ditangani dalam kehidupan sehari-hari. Begitu orang yang dicintai mencapai batas emosi dan fisik mereka, mereka harus membuat perubahan. Pengaturan waktu luang atau penggunaan dukungan psikoterapi dapat membantu untuk mengalami keseimbangan yang sesuai. Berurusan dengan penyakit seringkali membuat stres sehingga komplikasi psikologis dapat terjadi.
Rehabilitasi
Perawatan lanjutan untuk sindrom Smith-Magenis (SMS) pada dasarnya ditujukan untuk mengobati gejala penyakit. Karena SMS bersifat genetik. Penyembuhan tidak mungkin dilakukan. Cakupan perawatan dan pemeriksaan setelah perawatan bisa sangat kompleks dengan SMS.
Spektrum gejala yang mungkin terjadi berkisar dari kelainan fisik dan keterlambatan perkembangan (baik fisik dan mental) hingga gangguan perilaku yang parah, cacat intelektual dan penyakit serius pada organ dalam. Gejala yang ditimbulkan oleh SMS umumnya tidak kunjung sembuh. Tugas perawatan lanjutan adalah mencapai dan mempertahankan kualitas hidup setinggi mungkin bagi orang-orang dengan SMS.
Oleh karena itu, fokus perawatan setelah perawatan adalah melanjutkan intervensi terapeutik. Sebagai hasil dari perawatan lanjutan, anak-anak dan dewasa muda yang menderita SMS pasti telah belajar secara khusus untuk hidup dengan gejala penyakit ini sebaik mungkin. Selain pengobatan obat seumur hidup, dokter juga meresepkan pengobatan dan terapi wicara, terapi okupasi dan terapi musik sebagai pengobatan lanjutan (diulang secara teratur).
Sebagai tindakan swadaya, kerabat dapat menciptakan lingkungan sosial yang stabil selama perawatan setelah orang yang menderita SMS dan secara optimal menyusun kehidupan sehari-hari untuk kepentingan orang yang sakit. Jika gejala Anda memburuk, Anda mungkin memerlukan pemeriksaan lanjutan dengan dokter spesialis.
.jpg)
.jpg)

.jpg)


.jpg)