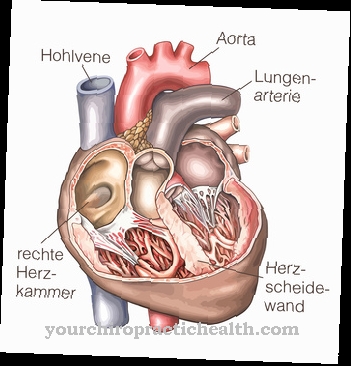
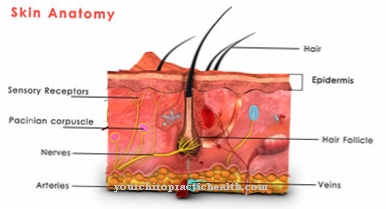
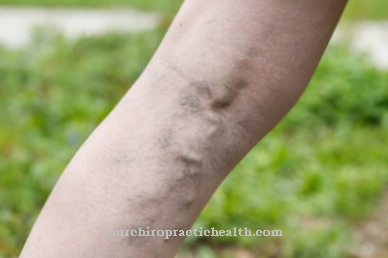
Di mana Sindrom Ehlers-Danlos, juga dikenal sebagai EDS, adalah kelainan jaringan ikat yang diturunkan sebagai bagian dari cacat genetik. Yang terpenting, EDS memanifestasikan dirinya melalui persendian yang hipermovable serta kulit yang terlalu meregang. Kadang-kadang, pembuluh darah, ligamen, otot, tendon, dan organ dalam juga dapat dipengaruhi oleh EDS; prognosisnya tergantung pada jenis sindrom yang didiagnosis.
Apa itu Sindrom Ehlers-Danlos?

© Astrid Gast - stock.adobe.com
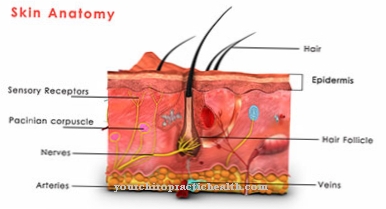
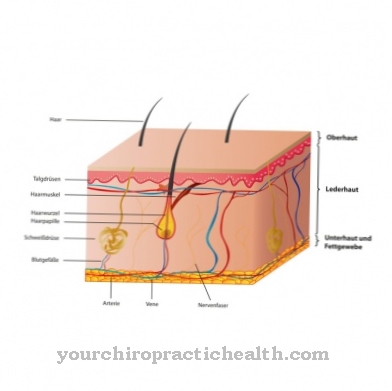
Sindrom Ehlers-Danlos adalah penyakit jaringan ikat yang sangat langka. Ini adalah penyakit bawaan yang terjadi sebagai bagian dari cacat genetik. Ada gangguan sintesis kolagen. Karena jaringan ikat terdapat di seluruh tubuh, gejala dan tanda sindrom Ehlers-Danlos bisa sangat berbeda dan bervariasi.
Kadang-kadang orang yang terkena dapat mengeluhkan kulit yang terlalu meregang; dalam beberapa kasus bahkan ada pembicaraan tentang pecahnya pembuluh darah dan organ dalam. Ada sepuluh jenis EDS, yang memiliki gejala dan jalur berbeda.
penyebab
Sindrom Ehlers-Danlos dipicu oleh kerusakan genetik pada sintesis kolagen. Sekitar 80 persen dari semua orang sakit menderita tipe I sampai III, tipe IV sampai VII hanya sebagian kecil dari semua orang sakit; Tipe VIII hingga X bahkan lebih jarang. Sindrom Ehlers-Danlos tipe I dan II memengaruhi kolagen V, sedangkan tipe III adalah kelainan kolagen III.
Gen berikut dapat memicu sindrom Ehlers-Danlos: COL1A1 hingga COL3A1 serta COL5A1 dan COL5A2 dan TNXB. Terkadang gen ADAMTS2 dan PLOD1 juga bertanggung jawab atas sindrom ini. Tergantung pada jenisnya, mode pewarisan juga tergantung. Banyak bentuk sindrom Ehlers-Danlos diturunkan sebagai sifat dominan autosomal.
Ini berarti bahwa hanya satu dari alel yang harus dipengaruhi oleh perubahan sindrom Ehlers-Danlos untuk pecah. Tetapi ada juga bentuk yang diwariskan sebagai sifat resesif autosom. Kedua alel harus menunjukkan perubahan agar penyakit dapat menular.
Gejala, penyakit & tanda
Karena gangguan sintesis kolagen, pembuluh darah, kulit dan persendian tidak berkembang dengan baik. Ini berarti bahwa pasien menderita kekurangan ketegasan dan, sebagai akibatnya, jaringan ikat yang terlalu meregang. Ini menyebabkan sedikit robeknya struktur yang sudah terpengaruh. Yang terpenting, pembuluh darah bisa robek, lengkungan tulang belakang atau usus pecah bisa terjadi. Kadang-kadang bahkan pneumotoraks berulang mungkin terjadi.
Dengan tipe I dan II, pasien mengeluhkan kulit yang sangat mudah terluka dan meregang. Proses penyembuhan luka sangat tidak normal dan persendiannya sangat kuat, dan dalam beberapa kasus pembuluh darah dan organ dalam juga dapat terpengaruh.
Pada tipe III, kulit hanya terpengaruh sebagian oleh kelainan; Yang terpenting, mereka yang terkena mengeluhkan terlalu banyak mobilitas sendi yang terlihat. Tipe IV memiliki kulit yang sangat tipis, hampir tembus cahaya. Pasien memiliki kecenderungan yang jelas untuk hematoma dan juga memiliki persendian yang sangat hipermovable, di mana pada tahap ini pembuluh dan organ dalam sering juga terkena sindrom Ehlers-Danlos.
Tipe VI terutama mengeluhkan kulit yang terlalu meregang. Penyembuhan luka tidak normal; terkadang organ dalam juga terlibat. Dalam banyak kasus, ada juga keterlibatan mata. Pada tipe VII A dan B, kulitnya terlalu meregang, yang terkadang sangat tipis. Banyak pasien juga mengeluhkan dislokasi pinggul. Sebaliknya, dengan tipe VII C, pasien melaporkan kulit kendur dan keterlibatan organ dalamnya. Sendi juga bisa digerakkan.
Diagnosis & kursus
Dokter membuat diagnosis klinis, terutama mengacu pada riwayat keluarga. Dokter juga memeriksa peningkatan kerapuhan kapiler menggunakan tes Rumpel-Leede dan juga melakukan biopsi kulit, yang dilanjutkan dengan pemeriksaan mikroskopis elektron pada seluruh struktur kolagen. Jenisnya dapat diidentifikasi menggunakan amplifikasi DNA genetik manusia.
Tergantung pada jenisnya, prognosis dan perjalanan penyakit sindrom Ehlers-Danlos bervariasi. Sementara beberapa pasien memiliki sedikit keterbatasan, yang lain memiliki masalah dalam kehidupan sehari-hari. Yang terpenting, rasa sakit, ketidakstabilan sendi yang ekstrem, dan deformasi tulang belakang berarti ada hambatan besar dalam mobilitas.
Mayoritas pasien, bagaimanapun, memiliki harapan hidup yang normal; Hanya pada sangat sedikit pasien yang juga menderita kerusakan pembuluh darah, komplikasi yang signifikan muncul dalam konteks sindrom Ehlers-Danlos.
Komplikasi
Komplikasi pada sindrom Ehlers-Danlos sangat bergantung pada jenis yang didiagnosis. Dalam kasus yang parah, kulit, otot, dan ligamen berubah bentuk. Organ internal juga bisa terpengaruh. Kulit pasien sering meregang dan persendian bisa bergerak berlebihan.
Anak-anak khususnya sangat menderita sindrom Ehlers-Danlos karena penindasan dan pelecehan. Hal ini dapat menyebabkan depresi dan masalah psikologis.
Kulit bisa rusak bahkan dengan sedikit pengaruh mekanis dan mudah robek. Ini meningkatkan risiko kecelakaan dan peradangan.
Tulang belakang juga dipengaruhi oleh sindrom Ehlers-Danlos, yang dapat menyebabkan tulang belakang melengkung. Dengan segala jenis penyembuhan luka terkadang tidak normal, sehingga infeksi dan radang harus ditangani dengan perawatan khusus. Pengobatan atau terapi tidak mungkin dilakukan dengan sindrom Ehlers-Danlos.
Namun, pasien harus berhati-hati untuk tidak melakukan aktivitas fisik yang berat. Demikian pula, luka dan pilek harus ditangani dengan sangat hati-hati.Sindrom Ehlers-Danlos tidak menyebabkan penurunan harapan hidup. Komplikasi dapat muncul terutama dari lengkungan tulang belakang atau penyembuhan luka yang tertunda.
Kapan sebaiknya Anda pergi ke dokter?
Berkonsultasi dengan dokter untuk mengetahui seringnya kulit robek, penyembuhan luka yang tidak normal, dan gejala lain yang menunjukkan kerusakan jaringan ikat. Sindrom Ehlers-Danlos juga dimanifestasikan oleh hematoma, kulit yang sangat sensitif dan terkadang juga dengan lengkungan tulang belakang dan ruptur usus. Lebih lanjut, masalah pinggul dan penyakit pada organ dalam juga bisa terjadi. Klarifikasi medis segera diperlukan untuk semua keluhan ini.
Karena sindrom Ehlers-Danlos adalah penyakit keturunan, masuk akal untuk melihat riwayat keluarga. Calon orang tua yang menderita penyakit itu sendiri atau memiliki kasus penyakit dalam keluarga mereka sebaiknya memeriksakan anak segera setelah lahir.
Paling lambat saat komplikasi serius seperti pendarahan atau nyeri hebat terjadi, dokter harus mengklarifikasi penyebabnya. Kontak lainnya adalah dokter kulit, berbagai internis atau spesialis penyakit keturunan. Dalam keadaan darurat medis, yang terbaik adalah segera memberi tahu layanan ambulans atau membawa orang yang terkena dampak ke rumah sakit terdekat.
Dokter & terapis di daerah Anda
Perawatan & Terapi
Tidak ada terapi simptomatik atau kausal. Untuk alasan ini, semuanya berputar di sekitar profilaksis dari setiap kerusakan konsekuensial. Pasien harus berhati-hati untuk menghindari cedera dan ketegangan fisik utama. Artinya, olahraga apa pun yang terkadang meningkatkan risiko cedera harus dihindari.
Risikonya meningkat selama kehamilan; Khususnya orang yang terkena tipe I, II, IV dan VI harus menjalani pemantauan ketat. Penting juga untuk memiliki terapi penekan batuk untuk pilek. Ini karena pecahnya usus besar dan selanjutnya pneumotoraks dapat dicegah. Jika terjadi luka, mereka harus dirawat dengan hati-hati untuk mendorong penyembuhan luka yang terganggu.
Outlook & ramalan
Prognosis sindrom Ehlers-Danlos (EDS) tergantung pada jenis penyakitnya. Ini juga berbeda untuk setiap orang yang terpengaruh. Pasien dengan tipe EDS hypermobile biasanya memiliki harapan hidup yang normal. Pada EDS tipe vaskular, ada risiko komplikasi yang lebih besar yang disebabkan oleh perdarahan internal mendadak. Namun, dalam kedua bentuk tersebut, kualitas hidup sangat terpengaruh.
Lebih lanjut, kedua bentuk sindrom Ehlers-Danlos mengandung beberapa penyakit keturunan pada jaringan ikat. Oleh karena itu, saat ini tidak ada terapi kausal yang memungkinkan. Bahkan pilihan untuk terapi simtomatik pun terbatas. Oleh karena itu penting bagi pasien untuk berhati-hati agar tidak mengalami cedera apapun. Bahkan operasi biasa tidak boleh dilakukan untuk menghindari cedera. Namun, ada pasien yang hampir tidak menderita keterbatasan. Yang lainnya mengalami gangguan parah dalam kehidupan sehari-hari mereka.
Secara keseluruhan, sindrom Ehlers-Danlos bersifat progresif. Dalam perjalanan hidup, batasan meningkat. Ketidakstabilan sendi, deformasi tulang belakang, dan nyeri hebat dapat terjadi, yang sangat mengurangi mobilitas. Dalam bentuk penyakit vaskular, ada juga risiko konstan bahwa pembuluh darah akan pecah, yang menyebabkan komplikasi yang mengancam jiwa. Konsekuensi terburuk dari EDS, bagaimanapun, adalah penurunan kualitas hidup yang parah karena berkurangnya mobilitas dan ketakutan konstan akan ruptur yang menyakitkan.
pencegahan
Tidak ada pencegahan. Terkadang hanya pemeriksaan sebagai bagian dari diagnosis pra-implantasi atau diagnosis badan kutub yang dapat mencegah pewarisan sindrom Ehlers-Danlos.
Rehabilitasi
Dengan sindrom Ehlers-Danlos, orang yang terkena biasanya memiliki sangat sedikit atau bahkan tidak ada tindakan lanjutan yang tersedia. Karena ini penyakit bawaan, maka tidak bisa diobati secara kausal, melainkan hanya berdasarkan gejala, sehingga tidak bisa sembuh total. Oleh karena itu, deteksi dini pada sindrom Ehlers-Danlos menjadi latar depan sehingga tidak ada komplikasi dan keluhan lebih lanjut bagi yang bersangkutan.
Semakin dini sindrom tersebut dikenali oleh dokter, semakin baik perjalanannya biasanya. Jika pasien ingin memiliki anak, penyuluhan herediter juga dapat dilakukan untuk mencegah sindrom ini diturunkan ke keturunan. Dalam kebanyakan kasus, harapan hidup pasien tidak terpengaruh secara negatif oleh sindrom ini.
Namun, mereka yang terkena dampak sangat bergantung pada bantuan teman dan keluarga dalam kesehariannya agar dapat menjalani kehidupan sehari-hari. Pembuluh darah juga harus diperiksa secara rutin agar tidak ada retakan yang dapat mengancam nyawa yang bersangkutan. Pada anak dengan sindrom Ehlers-Danlos, pendidikan komprehensif harus diberikan untuk menghindari kebingungan.
Anda bisa melakukannya sendiri
Pasien tidak dapat menyembuhkan sindrom Ehlers-Danlos dengan alat bantu sendiri. Penyakit keturunan dianggap tidak dapat disembuhkan. Namun, dalam kehidupan sehari-hari, berbagai tindakan dapat dilakukan untuk meringankan gejala. Pilihan suportif harus digunakan berulang kali, karena minimalisasi gejala secara permanen tidak dapat dicapai dengannya.
Kelemahan jaringan ikat dapat disembunyikan dengan potongan modis pada pakaian dan aksesori. Mengenakan pakaian dalam yang membentuk tubuh dan menstabilkan dan pada saat yang sama pakaian yang agak longgar membantu sehingga orang lain tidak dapat melihat gangguan jaringan ikat dalam kehidupan sehari-hari. Pijat, krim, atau mandi bergantian merangsang sirkulasi darah di kulit dan menopang tubuh. Kegiatan olahraga membantu membangun otot. Dengan cara ini, beberapa area kelemahan jaringan ikat dapat diminimalkan.
Orang tua dan dokter harus menjelaskan secara rinci tentang sindrom Ehlers-Danlos dan menjelaskan gejala serta perjalanan penyakitnya. Anak mendapat manfaat dari informasi sepenuhnya. Pertukaran digital dengan orang sakit lainnya juga dapat membantu dalam mengatasi penyakit dalam kehidupan sehari-hari. Kepercayaan pada kompetensi sendiri harus didukung dan didorong secara khusus sejak awal kehidupan anak. Memiliki harga diri yang kuat membantu anak memiliki kualitas hidup yang baik dengan sindrom tersebut.







.jpg)


.jpg)

.jpg)